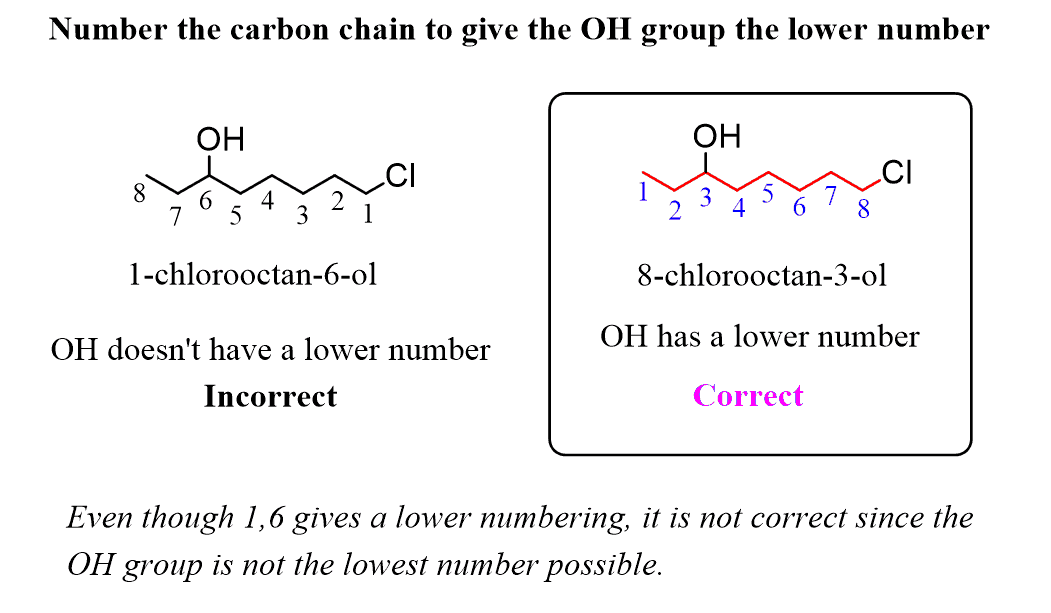
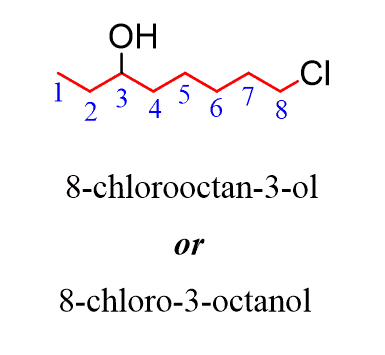
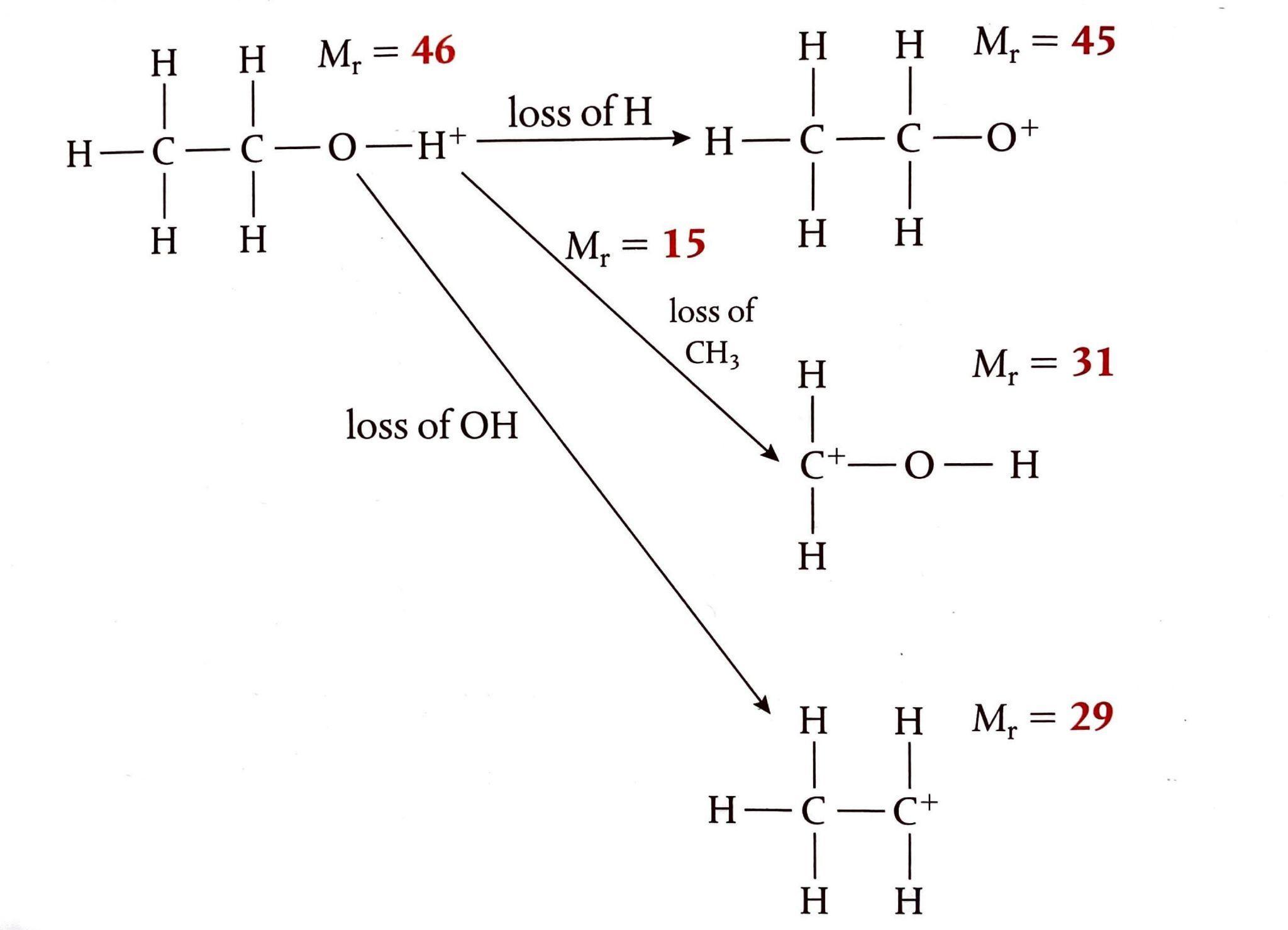
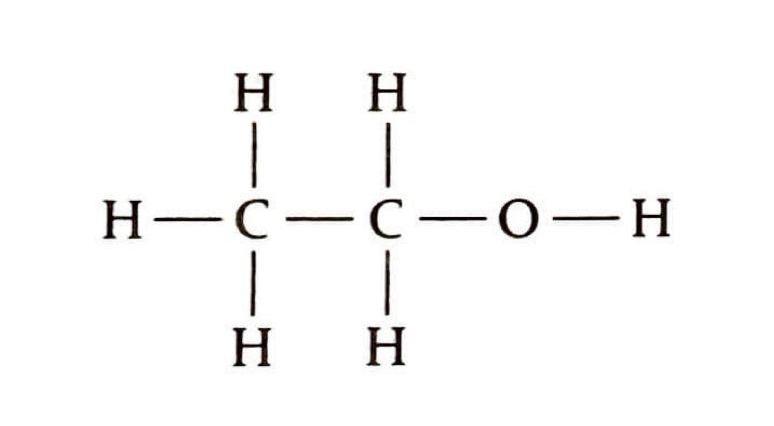
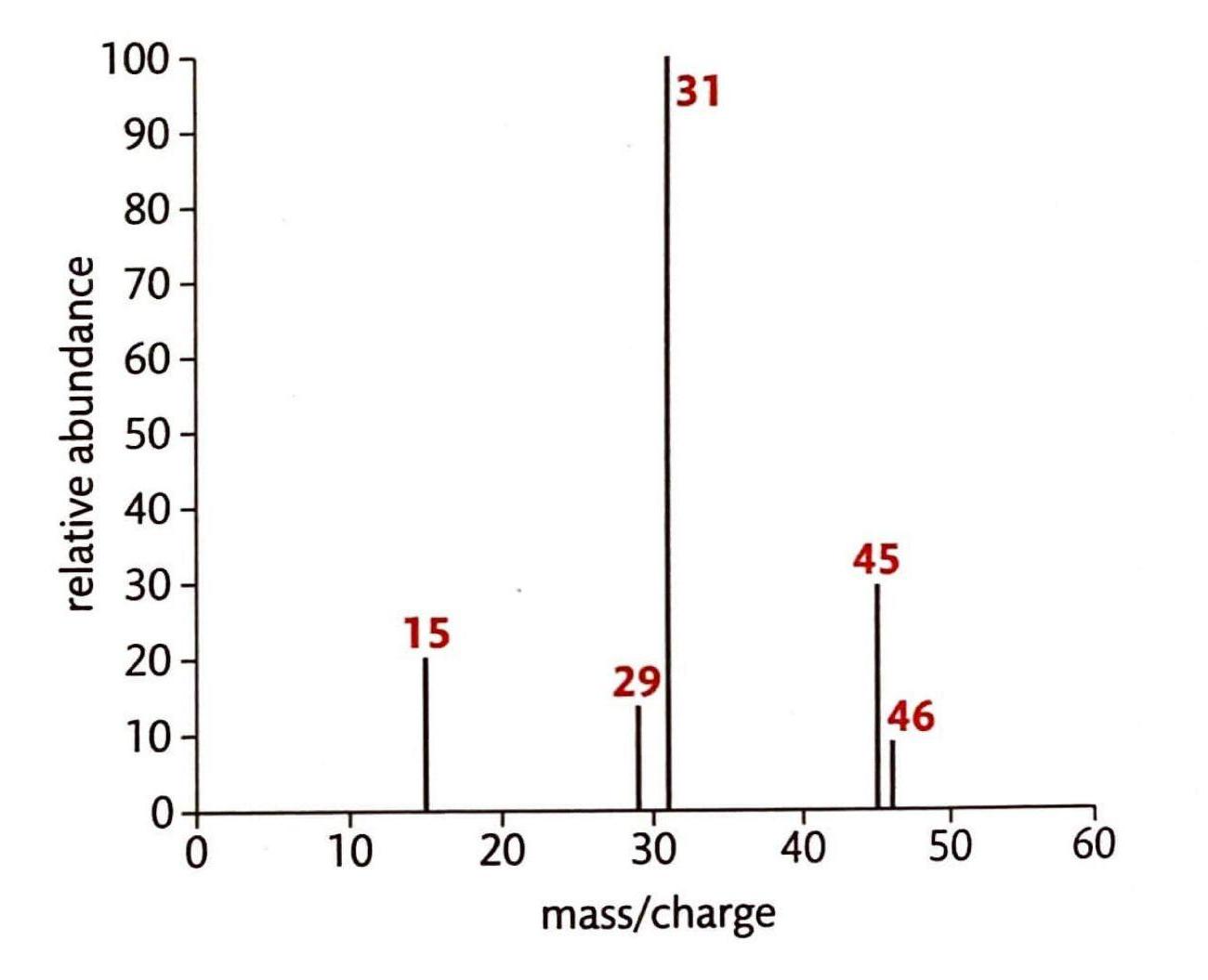
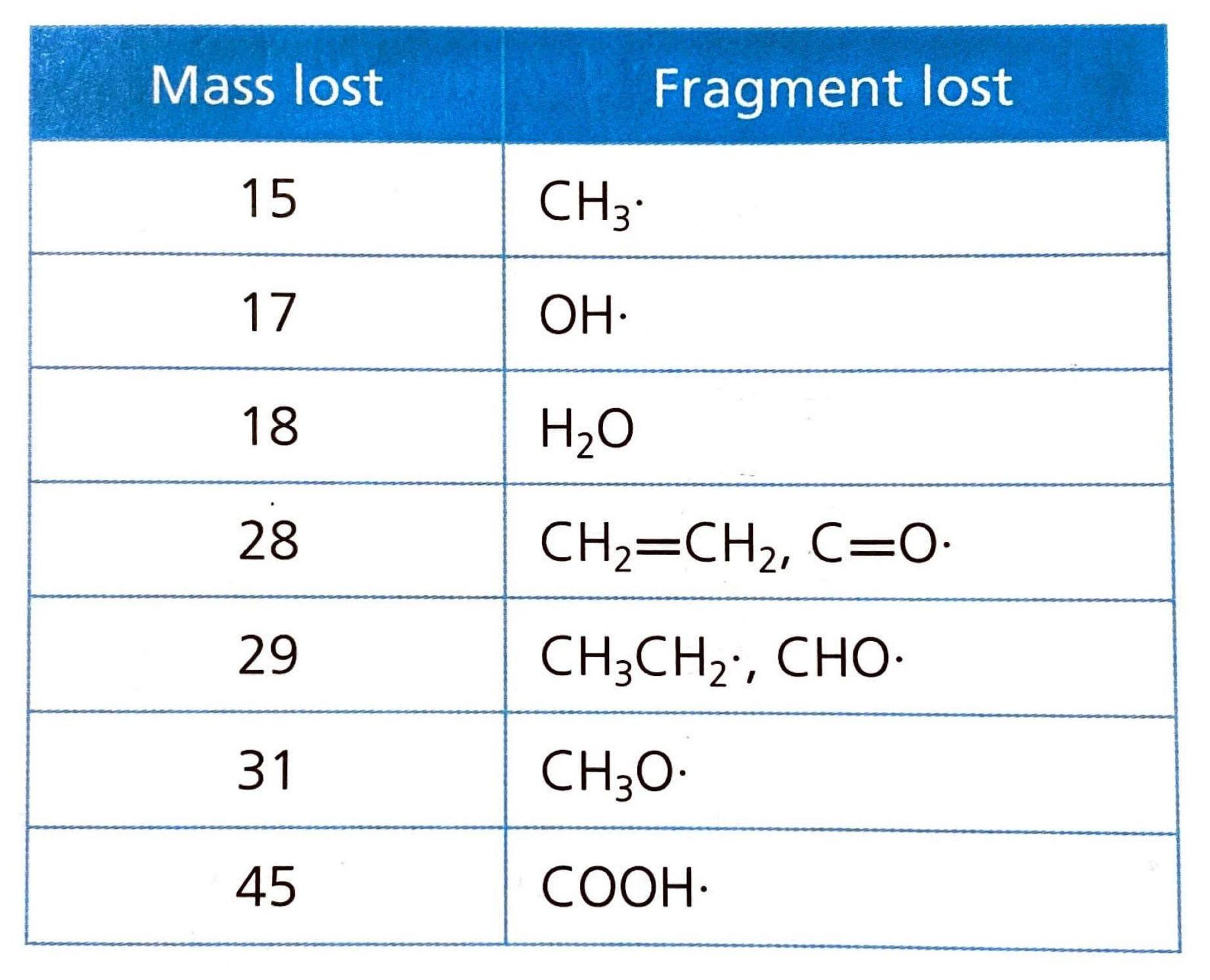
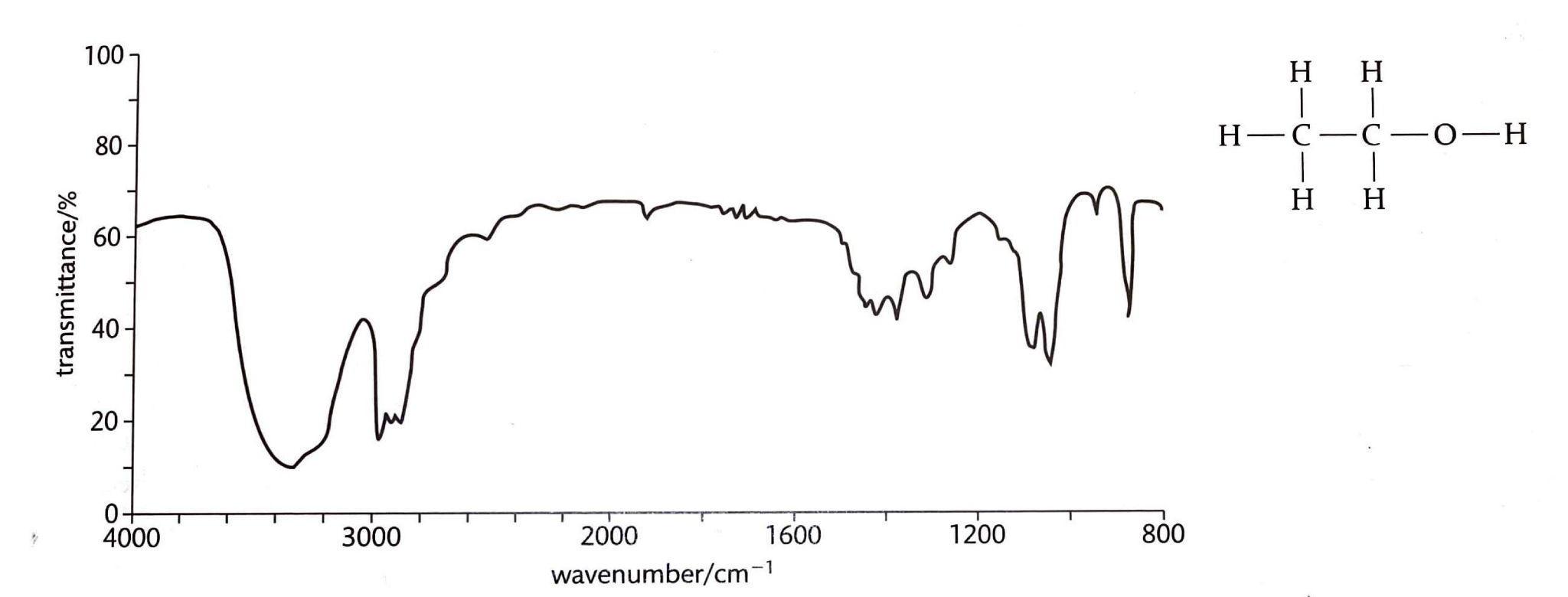
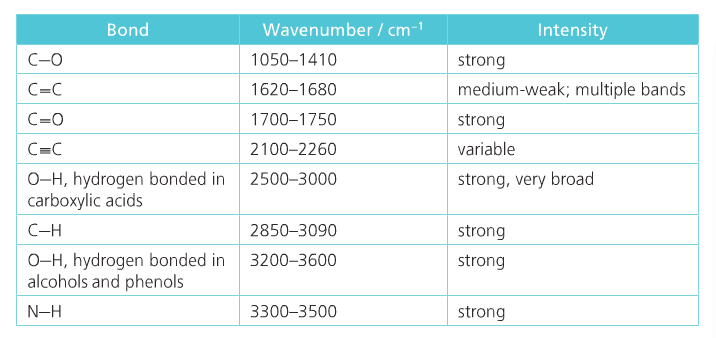
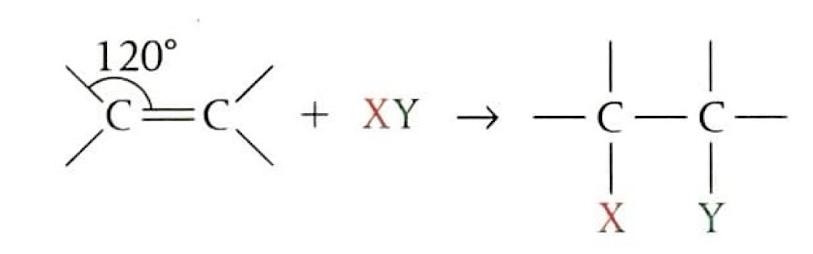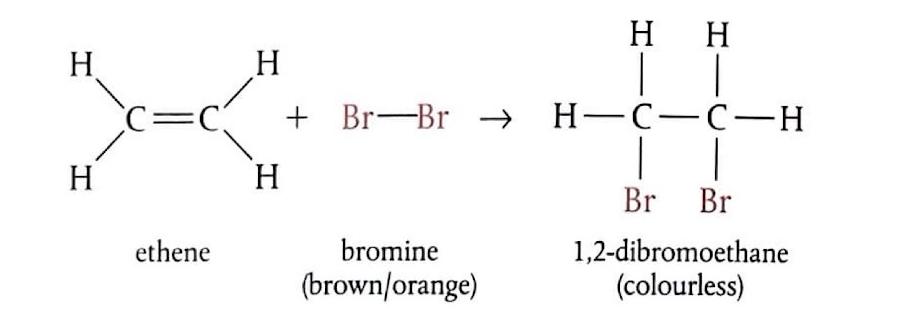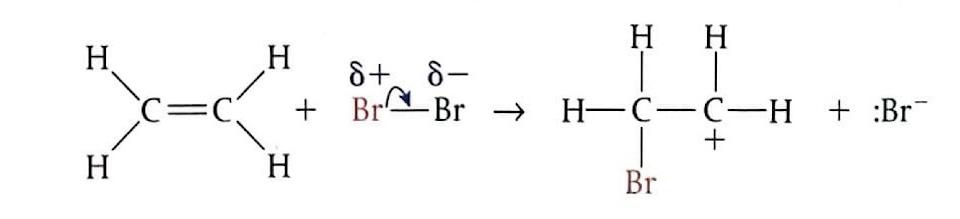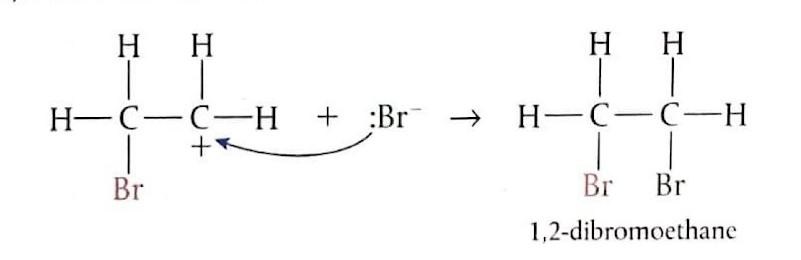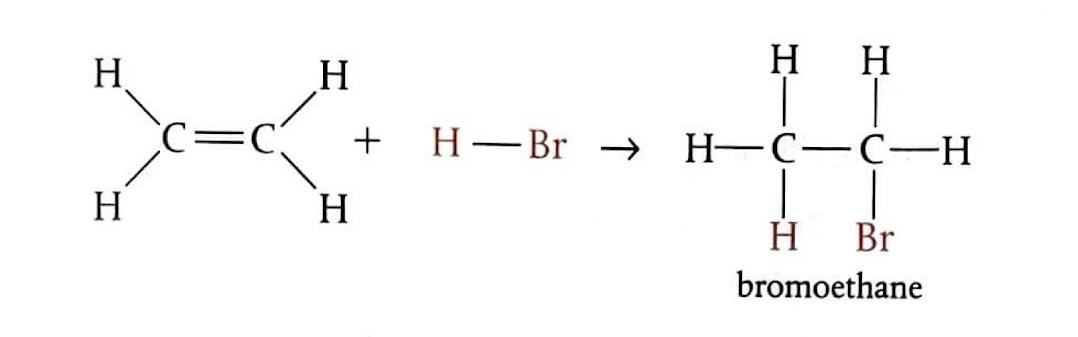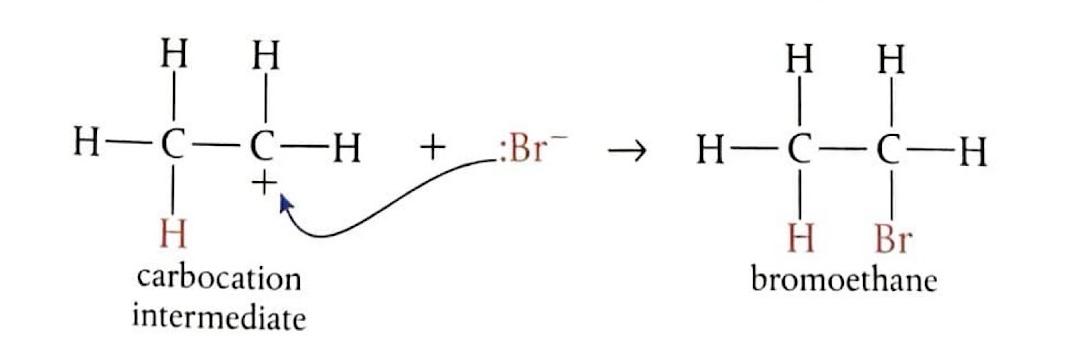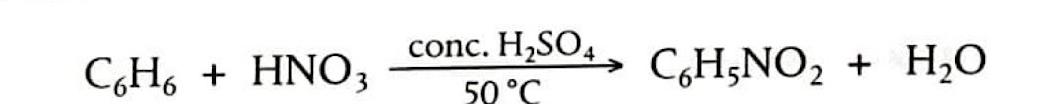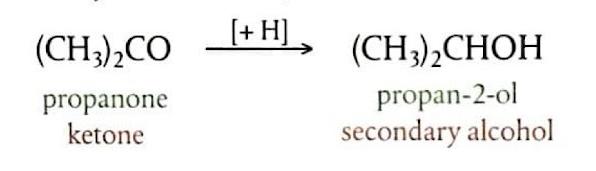Organic Reaction Classification
Organic reactions are systematically categorized based on both the transformation that occurs and the underlying mechanism by which it happens. This classification includes broad types such as nucleophilic substitution and electrophilic addition or substitution. A key distinction in these mechanisms lies in the nature of the reacting species: nucleophiles and electrophiles. Nucleophiles, acting as Lewis bases, are electron donors that form new covalent bonds. Common examples include hydroxide ions (OH
-), water (H
2O), ammonia (NH
3), and cyanide ions (CN
-). Conversely, electrophiles function as Lewis acids, accepting electron pairs. Examples of electrophiles include hydrogen ions (H
+), bromonium ions (Br
+), and nitronium ions (NO
2+).
Representing Electron Movement in Mechanisms
The movement of electron pairs during a reaction mechanism is visually represented by a curly arrow. The tail of this arrow indicates the origin of the electron pair, while the head points to its destination, illustrating the formation or breaking of a bond.
Nucleophilic Substitution (SN) Reactions
Nucleophilic substitution reactions frequently occur with halogenoalkanes. The carbon-halogen bond in these compounds is polar, rendering the carbon atom electron-deficient and thus susceptible to attack by a nucleophile. During the reaction, the carbon-halogen bond undergoes heterolytic fission, a process where both shared electrons in the bond are retained by the halogen atom. This results in the halogen being released as a halide ion (a negative ion), which is referred to as the leaving group.
Distinguishing SN1 and SN2 Reactions
Nucleophilic substitution reactions are broadly classified into two main types, S
N1 and S
N2, based on their reaction kinetics. S
N1 reactions are first-order, meaning their rate depends solely on the concentration of the substrate (the halogenoalkane). In contrast, S
N2 reactions are second-order, with their rate depending on the concentrations of both the substrate and the nucleophile.
Characteristics of SN2 Reactions
S
N2 reactions are single-step processes characterized by an unstable transition state. They are bimolecular, reflecting their second-order kinetics. In an S
N2 reaction, a strong nucleophile attacks the electrophilic carbon atom from the side opposite to the leaving group. This "backside attack" is crucial for the reaction's stereochemical outcome. S
N2 reactions proceed most rapidly with primary halogenoalkanes, where steric hindrance is minimized.
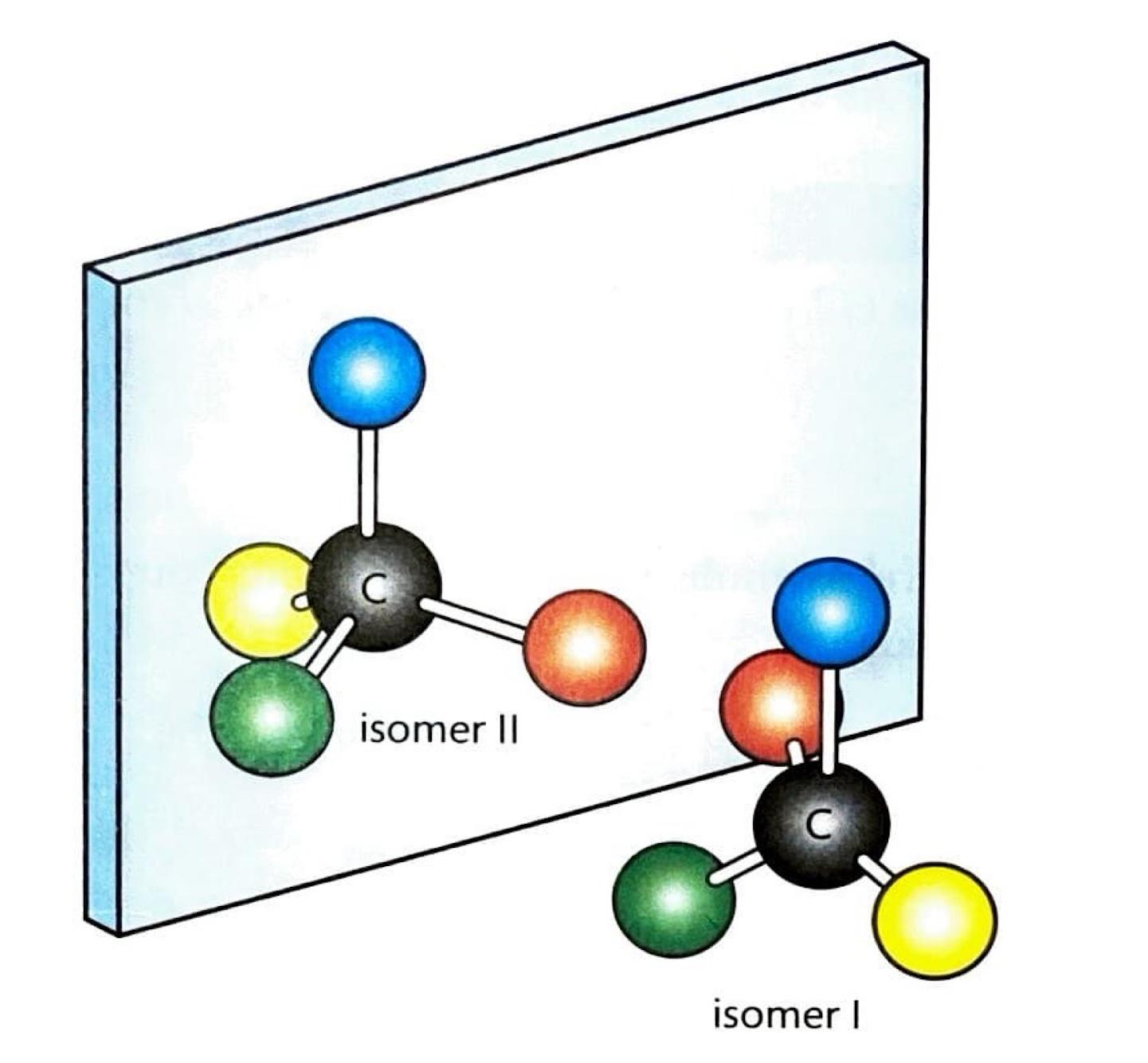
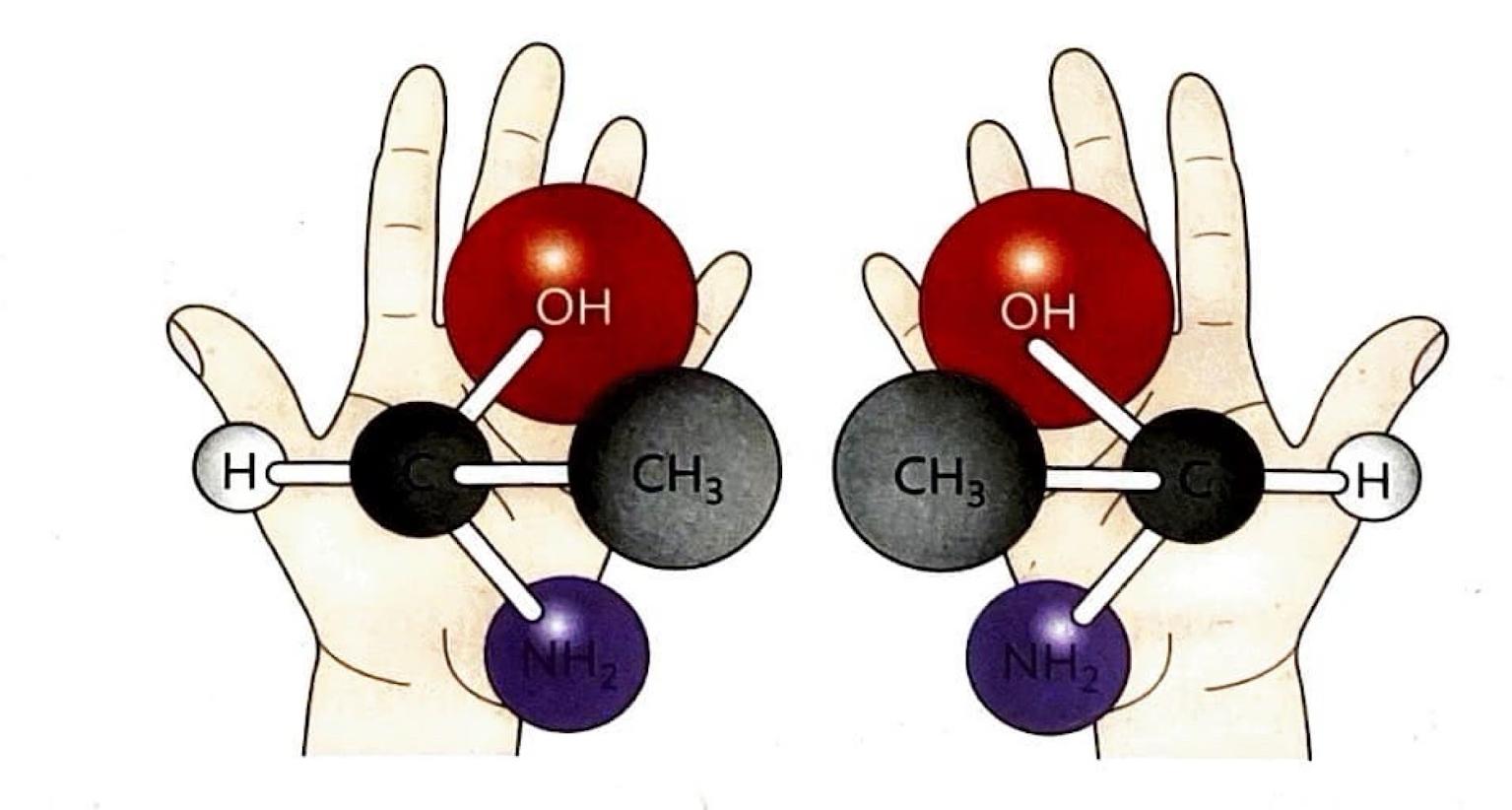
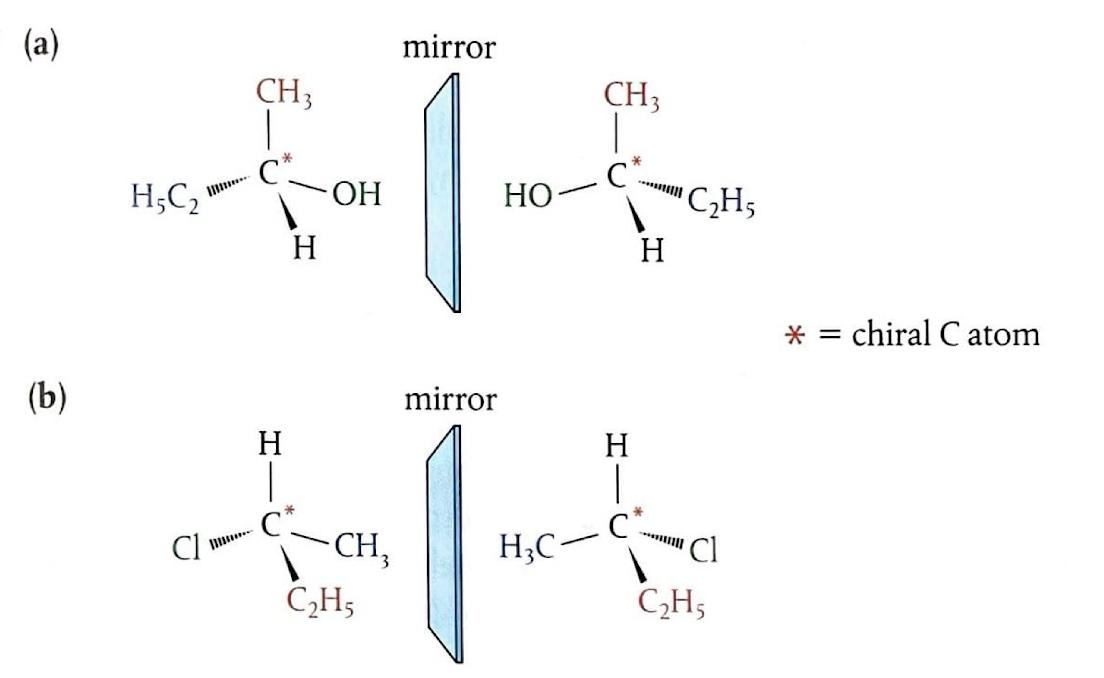
Stereochemistry in SN2 Reactions
A significant consequence of the backside attack in S
N2 reactions is the inversion of configuration at the carbon atom undergoing substitution. This means that the three-dimensional arrangement of groups around the carbon atom is inverted, much like an umbrella turning inside out.
S
N2 reactions are stereospecific, meaning that the stereochemistry of the reactants dictates the stereochemistry of the products. This is because bond formation and bond cleavage occur simultaneously in the transition state, preserving the stereochemical relationship. The precise three-dimensional arrangement of reactants directly determines the three-dimensional arrangement of the products, a characteristic that is particularly important in the synthesis of biologically active molecules such as amino acids and pharmaceuticals.
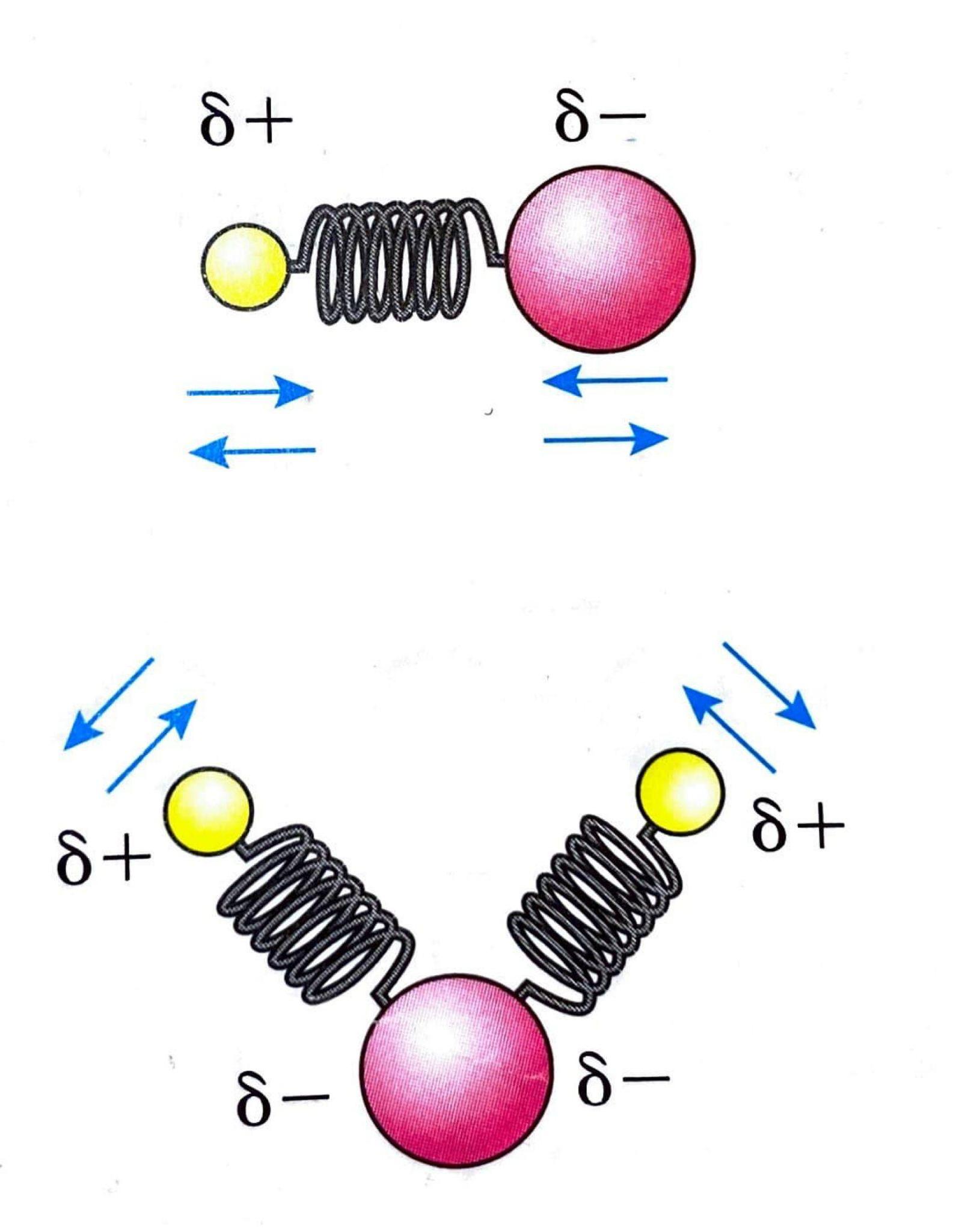
Solvent Effects on SN2 Reactions
S
N2 reactions are favored by polar, aprotic solvents. These solvents, such as propanone ((CH
3)
2CO), methylene chloride (CH
2Cl
2), and ethanenitrile (CH
3CN), lack -OH or -NH bonds and therefore do not form hydrogen bonds. They typically possess strong dipoles. Aprotic solvents solvate the metal cation associated with the nucleophile, effectively separating the nucleophile and increasing its energy state, which in turn enhances the reaction rate.
Characteristics of SN1 Reactions
S
N1 reactions are two-step processes that involve a carbocation intermediate. They are unimolecular, exhibiting first-order kinetics. In an S
N1 reaction, a relatively weak nucleophile can attack the electrophilic carbocation from either side, potentially leading to two different products. These reactions occur fastest with tertiary halogenoalkanes. The bulky alkyl groups in tertiary alkanes prevent direct nucleophilic attack via an S
N2 mechanism (steric hindrance). Furthermore, the three alkyl groups stabilize the carbocation intermediate through a positive inductive (electron-donating) effect, which helps to delocalize the positive charge.
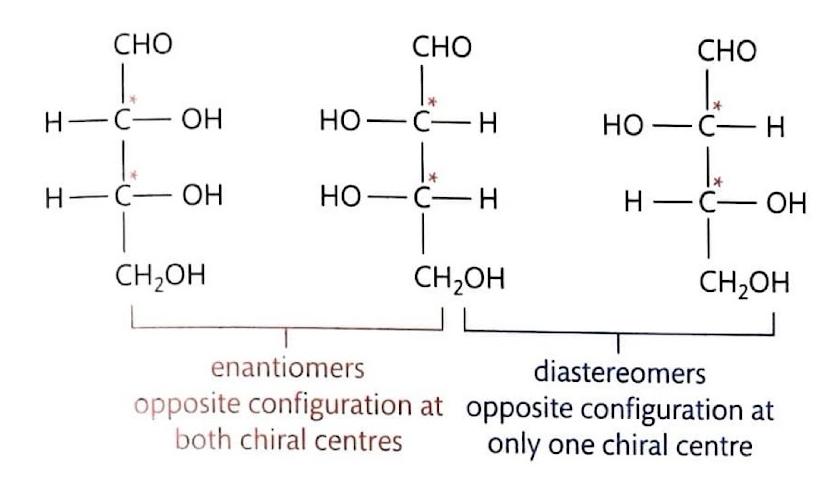
Stereochemistry in SN1 Reactions
Unlike S
N2 reactions, S
N1 reactions are not stereospecific. The carbocation intermediate, stabilized by inductive effects, persists long enough for the second step of the reaction to occur. Since the carbocation is planar, the nucleophile can attack from either face with equal probability, leading to a racemic mixture if the carbon is chiral.
Solvent Effects on SN1 Reactions
S
N1 reactions are favored by polar, protic solvents. These solvents, which include water, alcohols, and carboxylic acids, contain -OH or -NH bonds and are capable of forming hydrogen bonds. Protic solvents effectively stabilize the carbocation intermediate through ion-dipole interactions, thereby facilitating the S
N1 mechanism.
Nucleophilic Substitution in Secondary Halogenoalkanes
Secondary halogenoalkanes can undergo both S
N1 and S
N2 reactions, making it challenging to precisely predict the predominant mechanism of nucleophilic substitution without further information about the specific conditions.
Factors Influencing Nucleophilic Substitution Reaction Rates
The rates of nucleophilic substitution reactions are influenced by several factors, including the reaction mechanism (S
N1 vs. S
N2), the nature of the leaving group, and the solvent used.
Impact of Mechanism on Reaction Rates
S
N1 reactions generally proceed more quickly than S
N2 reactions, particularly in the presence of protic solvents. This enhanced rate is attributed to the ability of protic solvents to stabilize the carbocation intermediate formed in S
N1 reactions.
Influence of the Leaving Group on Reaction Rates
The nature of the leaving group significantly affects the rate of nucleophilic substitution. As one moves down a group in the periodic table, the electronegativity of the halogen decreases, making the carbon in the carbon-halogen bond less electron-deficient and thus less vulnerable to nucleophilic attack. However, the strength of the carbon-halogen bond also decreases down the group, making these bonds more easily broken. The overall effect on reaction rate depends on the balance of these factors. The reactivity of halogenoalkanes with silver nitrate in alcoholic solution can be used to compare the ease of C-X bond cleavage.
Influence of Solvent on Reaction Rates
As previously discussed, the choice of solvent plays a critical role in determining the favored mechanism and thus the reaction rate. S
N1 mechanisms are favored by polar, protic solvents, while S
N2 mechanisms are favored by polar, aprotic solvents. To experimentally track the rate of nucleophilic substitution reactions, silver nitrate can be added, and the formation of a precipitate of the silver halide (each having a distinct color) can be observed. For instance, to achieve the fastest nucleophilic substitution reactions, one would select conditions that optimize either the S
N1 or S
N2 pathway based on the substrate and desired outcome.
Electrophilic Addition Reactions
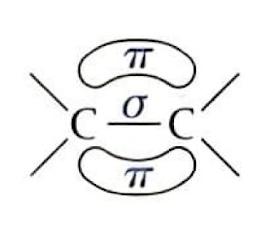
Alkenes are highly susceptible to electrophilic addition reactions due to the nature of their carbon-carbon double bond. This double bond consists of a strong sigma (σ) bond and a weaker pi (π) bond. The π bond, with its electron density located above and below the plane of the bond axis, is more easily broken and is highly attractive to electrophiles. The sp
2 hybridized carbon atoms of the double bond result in a trigonal planar geometry, an open structure that facilitates electrophilic attack. Examples of electrophilic addition include the addition of halogens and hydrogen halides to alkenes, which typically occur under mild conditions. In these reactions, the electrophile is often generated through heterolytic fission.
Electrophilic Addition of Bromine to Ethene
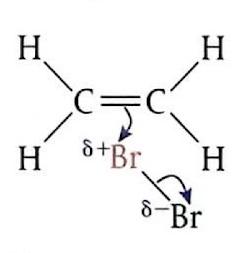
When ethene gas is bubbled through bromine water (which is brown) at room temperature, the product formed is 1,2-dibromoethane, which is colorless. The mechanism involves several steps: initially, the bromine molecule (Br
2) approaches the electron-rich region of the ethene double bond and becomes polarized. Subsequently, the Br
2 molecule undergoes heterolytic cleavage, and a bromonium ion (Br
+) attacks the alkene, breaking the π bond. This is the slow, rate-determining step and leads to the formation of an unstable carbocation intermediate. Finally, the bromide ion (Br
-) rapidly reacts with the carbocation intermediate to form the final product.
The formation of both BrH
2CCH
2Br and BrH
2CCH
2Cl from the reaction of ethene with Br
2 in the presence of Cl
- ions serves as evidence for the electrophilic addition mechanism, specifically the carbocation intermediate. If a carbocation is formed, it can react with any available nucleophile (Br
- or Cl
-), leading to a mixture of products.
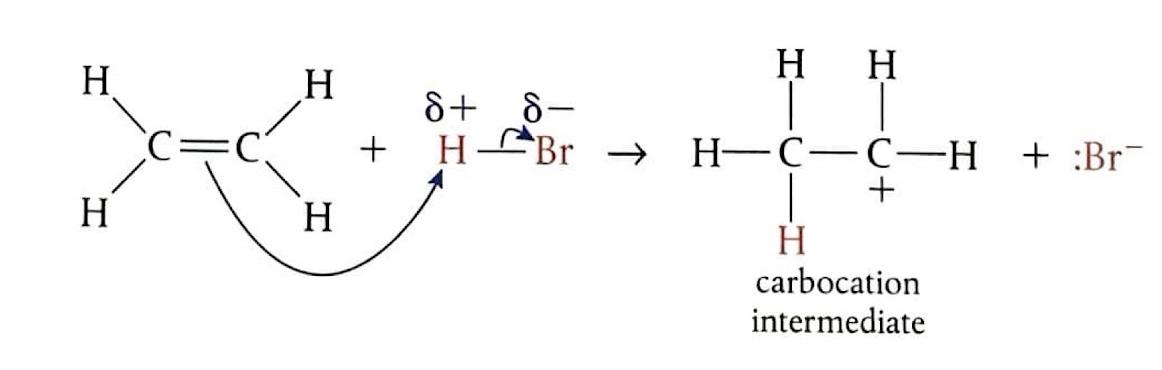
Electrophilic Addition of Hydrogen Bromide to Ethene
Bubbling ethene through concentrated hydrogen bromide (HBr) at room temperature produces bromoethane. In this reaction, HBr undergoes heterolytic fission to generate a hydrogen ion (H
+) and a bromide ion (Br
-). The electrophile, H
+, attacks the double bond of ethene, forming an unstable carbocation. This carbocation intermediate then quickly reacts with the Br
- ion to yield the final product.
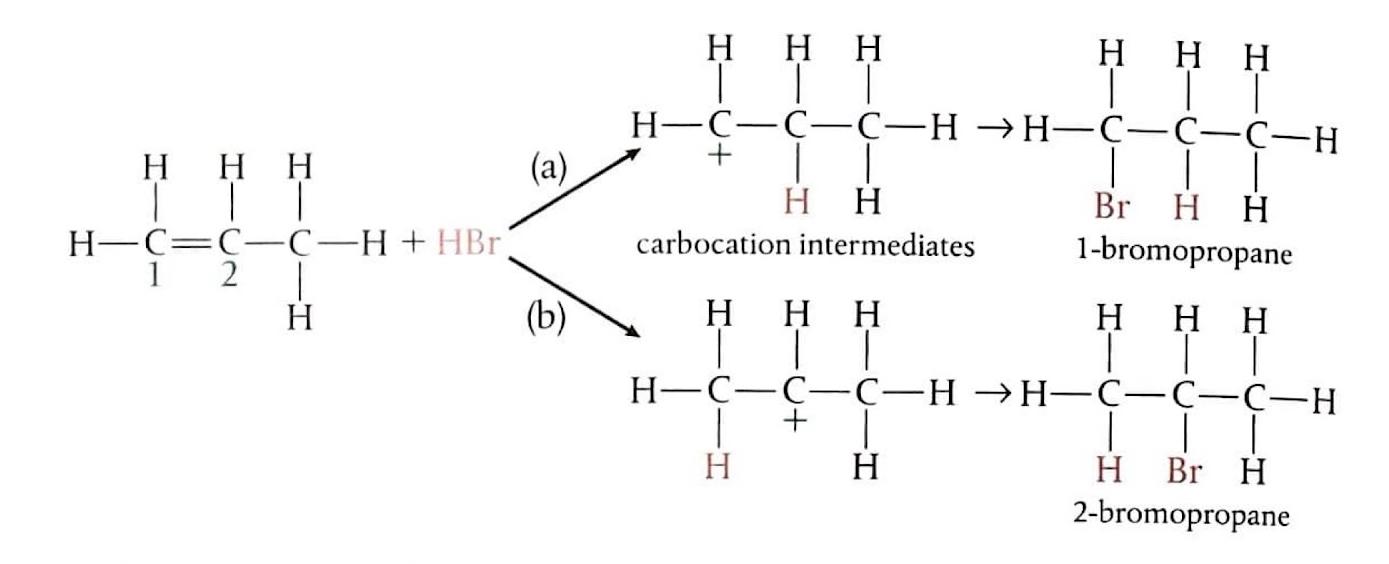
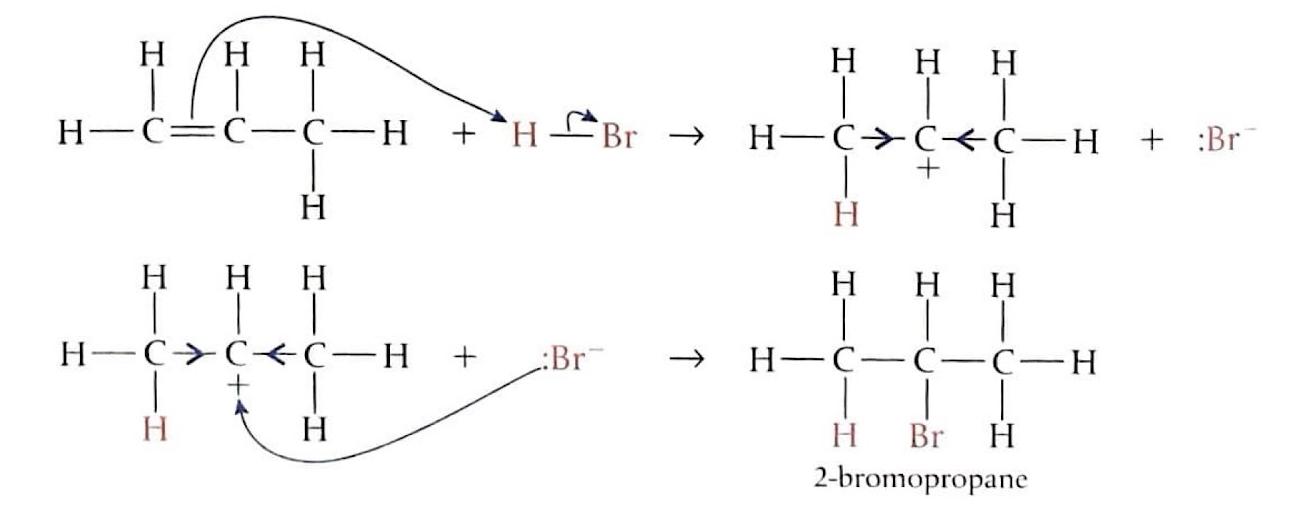
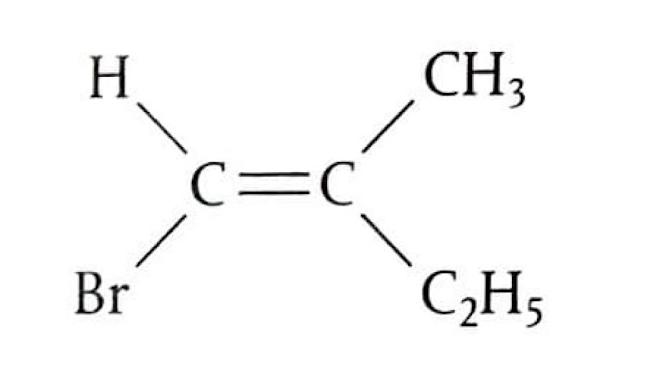
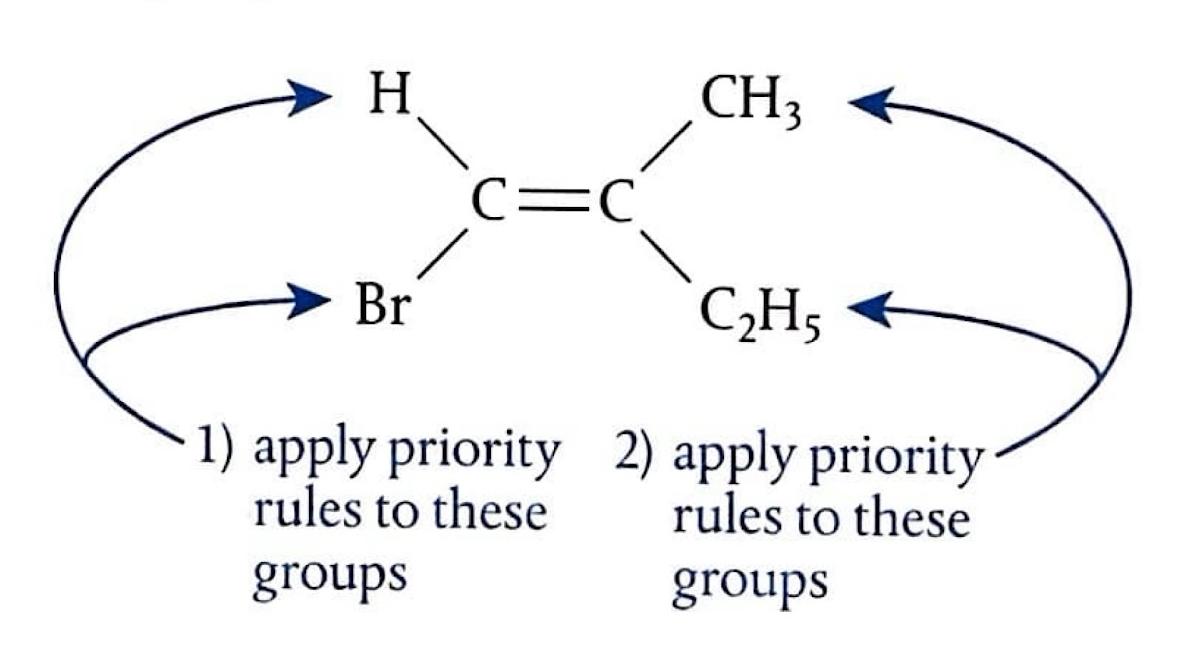
Electrophilic Addition to Unsymmetrical Alkenes: Propene + HBr
When an unsymmetrical alkene like propene reacts with a hydrogen halide such as HBr, two different products can theoretically form, depending on which carbon atom of the double bond the electrophile (H
+) bonds to initially.
In the case of propene and HBr, pathway (a) leads to the formation of a primary carbocation, while pathway (b) results in a secondary carbocation. The secondary carbocation is more stable due to the greater positive inductive effect from two alkyl groups compared to one in the primary carbocation. Consequently, 2-bromopropane is the main product formed, often referred to as the "major" product.
Markovnikov's Rule
Markovnikov's Rule provides a guideline for predicting the major product in electrophilic addition reactions to unsymmetrical alkenes. In technical terms, the more electropositive part of the reacting species (e.g., H
+ from HBr) bonds to the least highly substituted carbon atom in the alkene. In simpler terms, the hydrogen atom will attach to the carbon atom of the double bond that is already bonded to a greater number of hydrogen atoms. Conversely, the carbocation will form on the carbon atom bonded to more carbon atoms, as this leads to a more stable carbocation intermediate. When asked to predict the major products of an electrophilic addition reaction, it is crucial to explain the carbocation stability based on inductive effects.
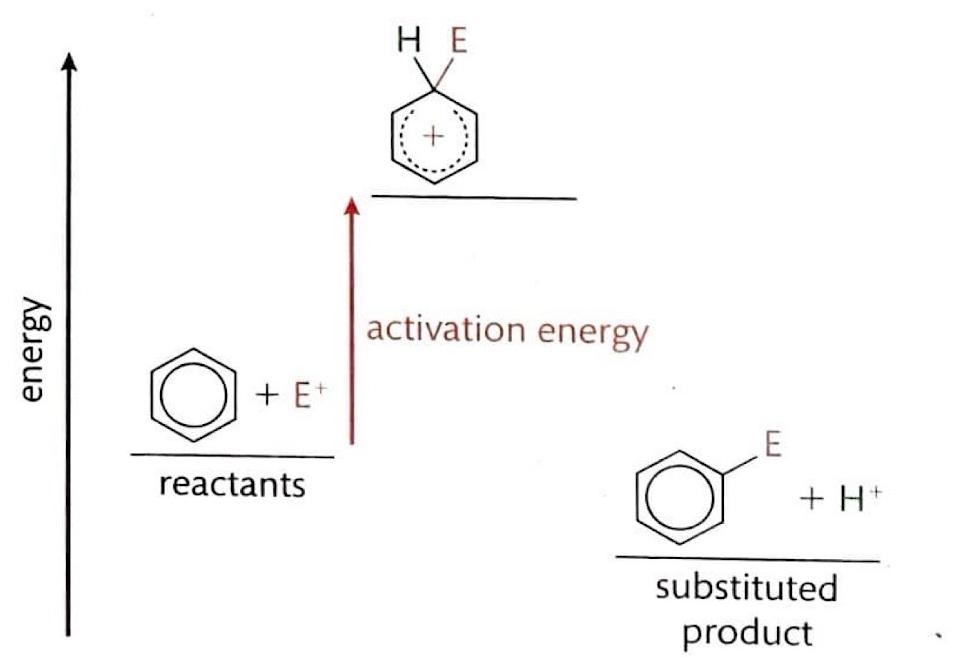
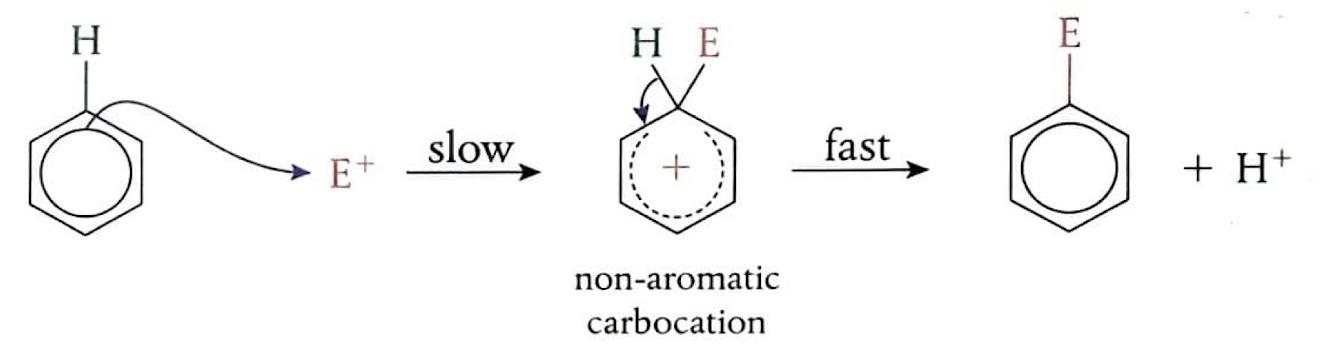
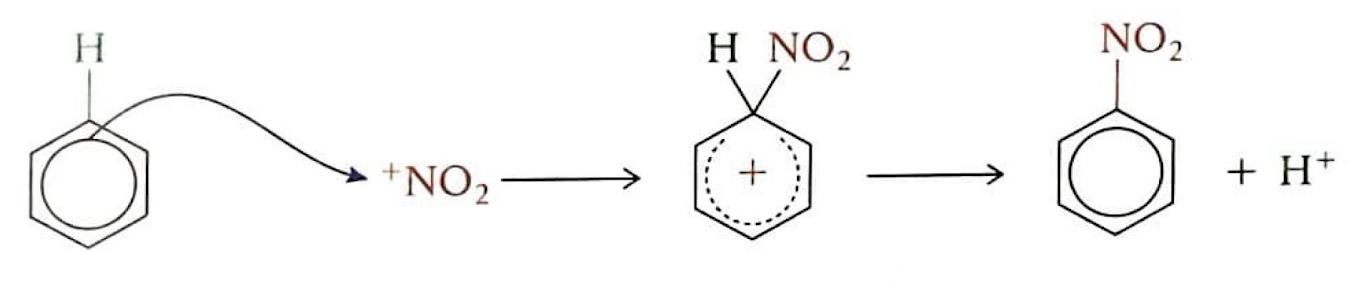
Electrophilic Substitution Reactions: Benzene
Despite being unsaturated, benzene exhibits a remarkably stable aromatic ring, which strongly favors substitution reactions over addition reactions. Benzene's delocalized π electron system creates a region of high electron density, making it attractive to electrophiles. In an electrophilic substitution reaction, a new bond is formed as one of the hydrogen atoms on the benzene ring is replaced. These reactions typically have a high activation energy and proceed slowly. The mechanism involves an electron pair from the benzene ring being attracted to the electrophile, which temporarily disrupts the delocalized π electron symmetry. This leads to the formation of an unstable carbocation intermediate where both the entering group and the leaving hydrogen are temporarily bonded. Subsequently, a hydrogen ion (H
+) leaves, restoring the aromaticity and forming a more stable, electrically neutral product.
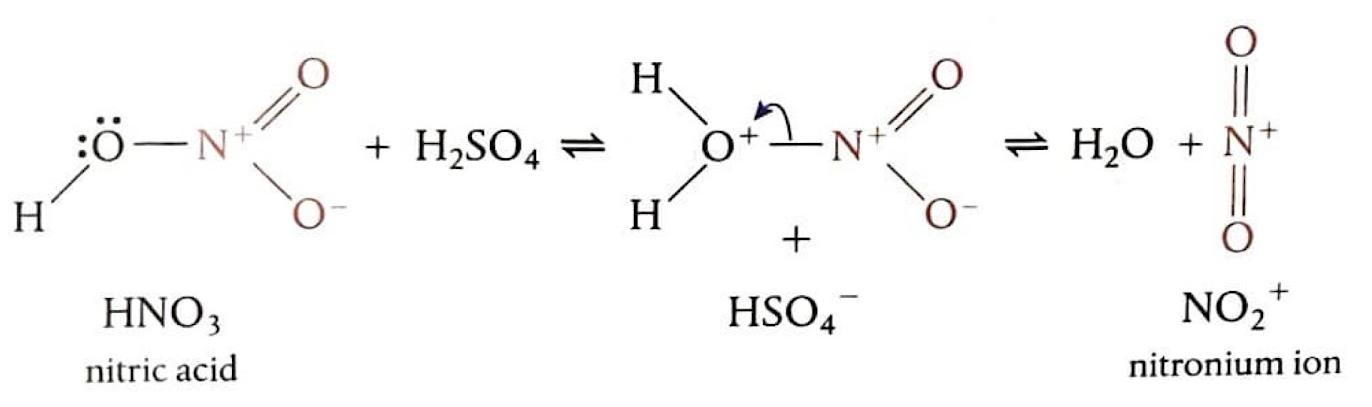
Electrophilic Substitution: Nitration of Benzene
The nitration of benzene is an electrophilic substitution reaction where a hydrogen atom on the benzene ring is replaced by a nitro group (-NO
2) to form nitrobenzene. The electrophile in this reaction is the nitronium ion (NO
2+), which is generated by mixing concentrated nitric acid and concentrated sulfuric acid at 50°C. Sulfuric acid acts as a catalyst by protonating nitric acid, leading to the formation of the highly electrophilic NO
2+ ion. This strong electrophile then attacks the benzene ring, forming a carbocation intermediate. Finally, a proton (H
+) is lost from the intermediate, which then binds to HSO
4- to regenerate the sulfuric acid catalyst, and the aromaticity of the ring is restored.
Reduction Reactions in Organic Chemistry
In organic chemistry, reduction is often characterized by a gain of hydrogen atoms or a loss of oxygen atoms.
Reduction of Carbonyl Compounds
The reduction of carbonyl compounds is essentially the reversal of oxidation reactions and can be achieved using specific reducing agents. Sodium borohydride (NaBH
4) is a common reducing agent used in aqueous or alcoholic solutions. For more powerful reductions, lithium aluminum hydride (LiAlH
4) is employed under anhydrous conditions, typically in dry ether followed by aqueous acid workup. Both NaBH
4 and LiAlH
4 function by producing a hydride ion (H
-), which acts as a nucleophile and attacks the electron-deficient carbonyl carbon. It is important to note that the reduction of carboxylic acids with strong reducing agents like LiAlH
4 cannot be stopped at the aldehyde stage due to the rapid further reduction of the aldehyde.
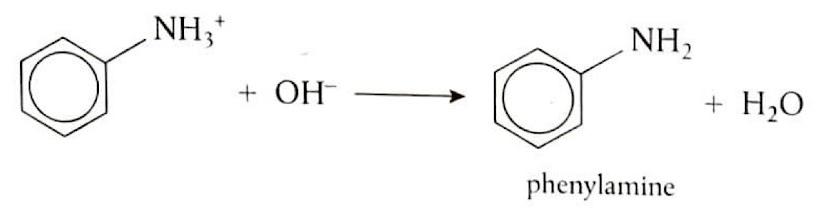
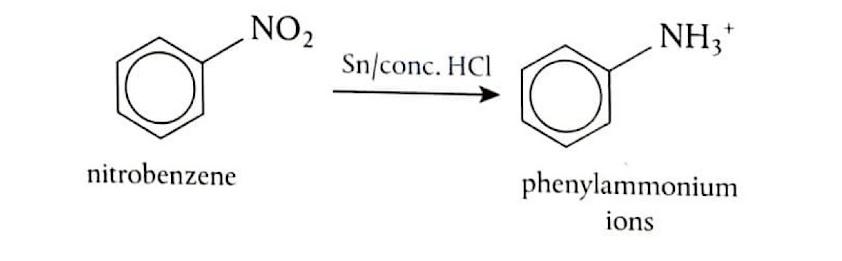
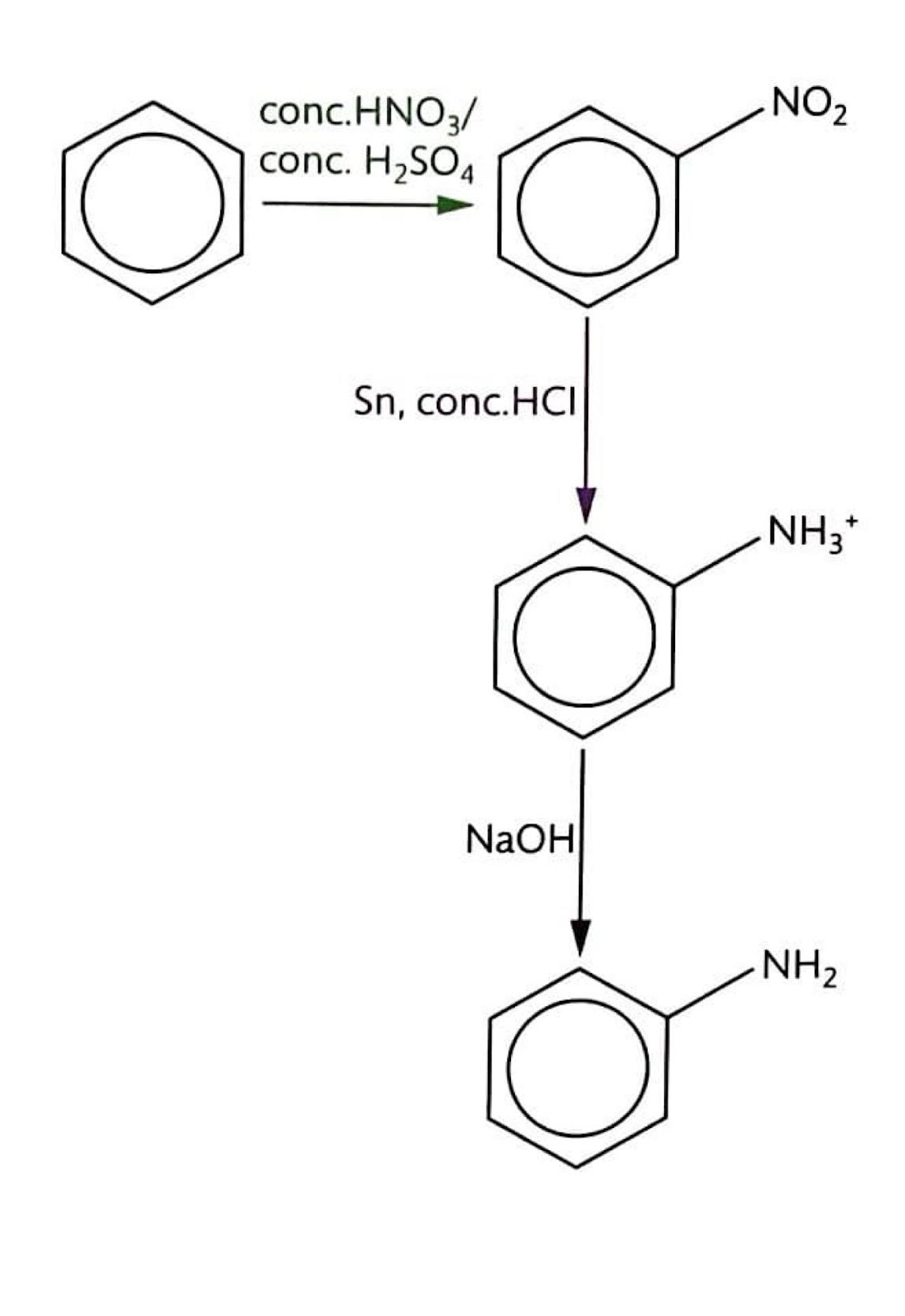
Reduction of Nitrobenzene
Nitrobenzene can be converted into phenylamine through a two-step reduction process. First, nitrobenzene (C
6H
5NO
2) is heated under reflux with tin (Sn) and concentrated hydrochloric acid (HCl). This reaction reduces the nitro group to an amino group, forming phenylammonium ions (C
6H
5NH
3+). In the second step, the phenylammonium ions are treated with a strong base, such as sodium hydroxide (NaOH), to deprotonate the ammonium group and yield phenylamine (C
6H
5NH
2).